![]()
![]()
Wednesday 3/29
7:00-8:30pm
WEL 2.224
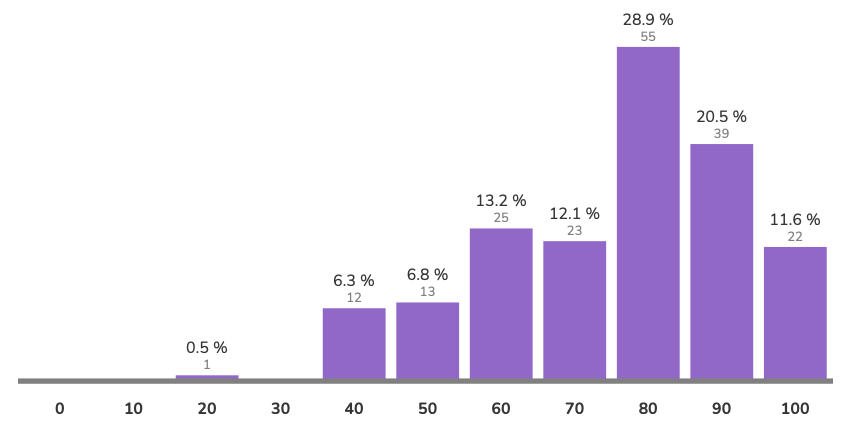
avg = 79.4
🔑 Here are all the KEYS to Exam 3
Students will be able to...
Students will be able to...
Pretty much all cations and anions in aqueous solutions are ±1, ±2, or ±3. There are really almost no charges above +3 or below -3. For that reason, the only salt ratios that we will encounter for MxXy type salts are 1 : 1 1 : 2 1 : 3 or 2 : 3 . And, since the cation / anion absolute charges could be reversed the flipped ratios are possible as well 2 : 1 3 : 1 or 3 : 2 .
| salt ratio | salt formula | aqueous cation | aqueous anion | equilibrium formula | Ksp(x) | molar solubility | ||
|---|---|---|---|---|---|---|---|---|
| 1:1 | MX(s) | ⇌ | M+(aq) | + | X–(aq) | Ksp = [M+][X–] | \(K_{\rm sp} = x^2\) | \(x = \sqrt{K_{\rm sp}}\) |
| 1:2 | MX2(s) | ⇌ | M2+(aq) | + | 2 X–(aq) | Ksp = [M2+][X–]2 | \(K_{\rm sp} = 4x^3\) | \(x = \root 3 \of {K_{\rm sp}\over 4}\) |
| 1:3 | MX3(s) | ⇌ | M3+(aq) | + | 3 X–(aq) | Ksp = [M3+][X–]3 | \(K_{\rm sp} = 27x^4\) | \(x = \root 4 \of {K_{\rm sp}\over 27}\) |
| 2:3 | M2X3(s) | ⇌ | 2 M3+(aq) | + | 3 X2–(aq) | Ksp = [M3+]2[X2–]3 | \(K_{\rm sp} = 108x^5\) | \(x = \root 5 \of {K_{\rm sp}\over 108}\) |
and the "flipped" ratios...
| salt ratio | salt formula | aqueous cation | aqueous anion | equilibrium formula | Ksp(x) | molar solubility | ||
|---|---|---|---|---|---|---|---|---|
| 2:1 | M2X(s) | ⇌ | 2 M+(aq) | + | X2–(aq) | Ksp = [M+]2[X2–] | \(K_{\rm sp} = 4x^3\) | \(x = \root 3 \of {K_{\rm sp}\over 4}\) |
| 3:1 | M3X(s) | ⇌ | 3 M+(aq) | + | X3–(aq) | Ksp = [M+]3[X–] | \(K_{\rm sp} = 27x^4\) | \(x = \root 4 \of {K_{\rm sp}\over 27}\) |
| 3:2 | M3X2(s) | ⇌ | 3 M2+(aq) | + | 2 X3–(aq) | Ksp = [M2+]3[X3–]2 | \(K_{\rm sp} = 108x^5\) | \(x = \root 5 \of {K_{\rm sp}\over 108}\) |
We can measure the rate of a reaction via any of the species in the over reaction:
\(a {\rm A} + b {\rm B} \rightarrow c {\rm C} + d {\rm D}\)
The rate of the "overall reaction as balanced" means you are measuring the rate of entire reactions as written. So each reactant and product is running at a scaled up version of the overall reaction. The "scaled up" amount is the coefficient for that species. For example, reactant A is running "a" times faster than the overall reaction. So to get ALL species to match up, we normalize the rates to the rate of one mole of reaction (the whole thing).
rate of reaction = \(-{\Delta[{\rm A}]\over a\Delta t} = -{\Delta[{\rm B}]\over b\Delta t} = {\Delta[{\rm C}]\over c\Delta t} = {\Delta[{\rm D}]\over d\Delta t}\)
Make sure you can convert the rate of any one species into the rate of another species. This is really just saying to know how stoichiometry works when dealing with rates.
Rates of reactions are dependent on the concentrations of the reactant (and sometimes product) species. This is generalized by showing the rate law for a reaction of A, B, and C going to products.
\({\rm rate\;of\;rxn} = k [{\rm A}]^x[{\rm B}]^y[{\rm C}]^z\cdots \)
\(x\) is the order in A, \(y\) is the order in B, and \(z\) is the order in C. Sum those orders all together and you'll have what is called the overall order. The overall order is important to know and the units for the rate constant, \(k\), depends on the overall order.
| name | zero order | first order | second order |
|---|---|---|---|
| rate law | \({\rm rate} = k\) | \({\rm rate} = k[{\rm A}]\) | \({\rm rate} = k[{\rm A}]^2\) |
| integrated rate law |
\([{\rm A}]_0 - [{\rm A}] = kt\) | \(\ln[{\rm A}]_0 - \ln[{\rm A}] = kt\) \(\ln\left({[{\rm A}]_0 \over [{\rm A}]}\right) = kt\) |
\({1\over[{\rm A}]} - {1\over[{\rm A}]_0} = kt\) |
| straight line form (\(y = mx + b\)) |
\([{\rm A}] = -kt + [{\rm A}]_0 \) | \(\ln[{\rm A}] = -kt + \ln[{\rm A}]_0 \) | \({1\over[{\rm A}]} = kt + {1\over[{\rm A}]_0}\) |
| half life | \(t_{1/2} = {[{\rm A}]_0\over 2k}\) | \(t_{1/2} = {\ln 2\over k}\) | \(t_{1/2} = {1\over [{\rm A}]_0 k}\) |
| to actually plot on x,y graph \(y\) = conc \(x\) = time |
\(y = -kx + {\rm A}_0 \) | \(y = {\rm A}_0 e^{-kx} \) | \(y = \left(kt + {1\over{\rm A}_0}\right)^{-1}\) |
Arrhenius Equation: \(k = A\,e^{-E_{\rm a}/RT} \)
\(E_{\rm a}\) is the activation energy for the reaction and it is always positive. The \(A\) is known as the Arrhenius "pre-exponential factor" and is unique for every reaction - it has the same units as the rate constant, \(k\). The factor can be eliminated by setting a ratio for two values of k at two different temperatures. Dr. McCord calls this the "user friendly" version of the Arrhenius equation.
\[\ln\left({k_2\over k_1}\right) = {E_{\rm a}\over R}\left({1\over T_1} - {1\over T_2}\right)\]
Notice that this is the third time you've seen this basic formula in CH302. The first time, it was the Clausius-Clapeyron equation for vapor pressures at two temperatures. The second time, it was the van't Hoff equation for the equilibrium constant, K, at two temperatures. Now the same set up works again but for rate constants. You should now be a "pro" at using this type of formula.
One more thing... the Arrhenius equation can be rewritten to be in a "straight line plot" form, \(y=mx+b\). That form is the following
\(\ln k = {-E_{\rm a}\over R}\left({1\over T}\right) + \ln A \)
So your plot is ln(k) vs 1/T and the slope will be equal to –Ea/R, and the y-intercept is equal to ln(A).